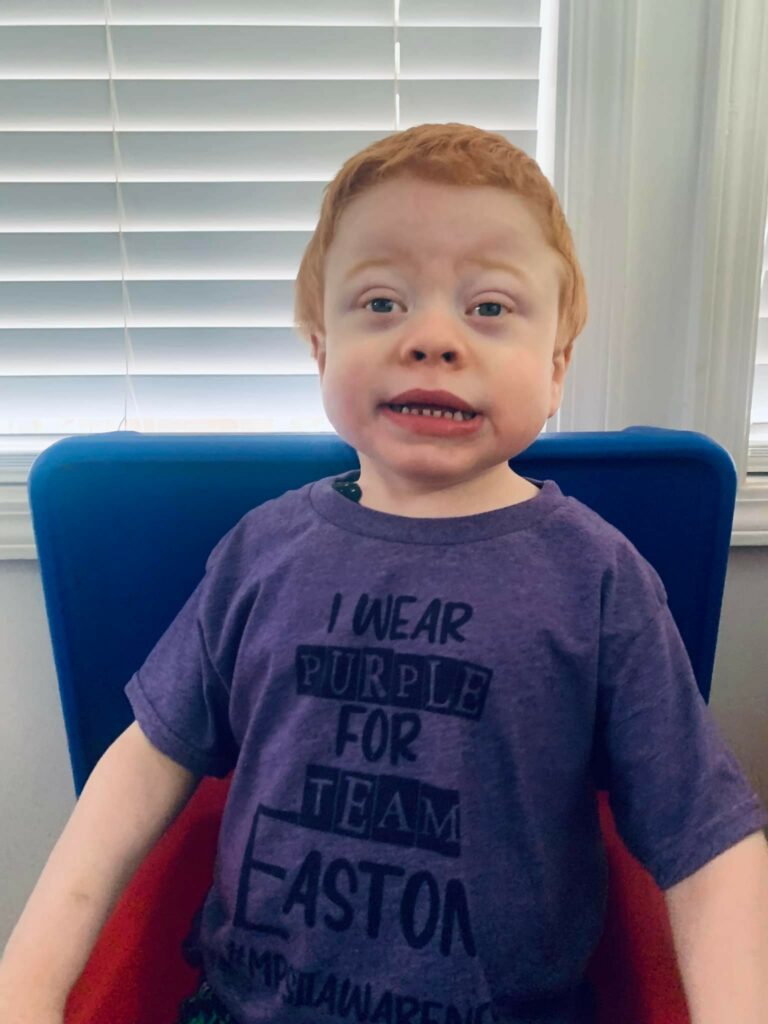
Today is MPS/ML Awareness Day and we wear purple to honor Easton, his brothers and sisters with MPS/ML and those who have lost their battle. All of our children need a cure. We need to spread awareness for this awful disease. We all want more time with our babies. 1 in 150,000. We enjoy the support and love our community has shown and we need you all to come together to save lives. I invite you to read Easton’s story to learn more about MPS.
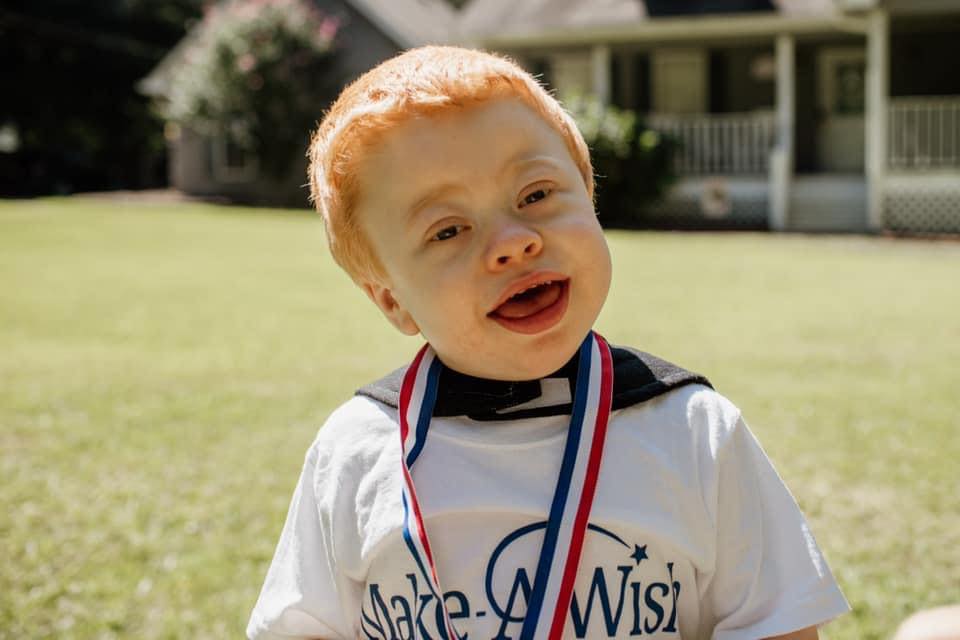
Easton was born at thirty-six weeks gestation on January 6, 2017. He was almost immediately taken to the NICU for something called PPHN (Persistent Pulmonary Hypertension), Easton was intubated for twelve days on a high-frequency ventilator, then C-PAP and oxygen until he was twenty-five days old. We were able to bring him home on day thirty-one. About two weeks after we brought Easton home, we rushed him to our local children’s hospital for what we came to find out was aspiration pneumonia. He was put on thickened feeds through his bottle and we thought all would be okay. Until the noisy breathing and breathing concerns persisted, among other things. We saw ENT, Pulmonology, Cardiology, Genetics, GI, Neurology, Plastics, General Surgery, Orthopedics and a Sleep Specialist. Everyone knew there was probably a bigger picture with his symptom list being a mile long, but nobody could really fit everything together. Easton had tests, procedures, bronchoscopy’s, sleep studies, etc. Some of which showed abnormalities and some that were no help. I demanded people make Easton a priority. It wasn’t until Easton was seen by his Orthopedic doctor to be casted, as he has a condition called Metatarsus Adductus, that really raised eyebrows. We questioned Easton’s spine months prior, his MRI showed some abnormalities, but it was not anything that we were really talked too about. However, his Orthopedic doctor ordered an x-ray and after showing it to a spine specialist- we heard the word, Mucopolysaccharidoses for the first time. I immediately went home and did what they tell you not to do, I researched. I questioned some things and thought they fit but then I saw the words: TERMINAL, LIFE-LIMITING. I automatically turned away from this and thought, there was no way. Not my family and not my son. A few weeks later after some other genetic tests came back and were normal, we decided to do the simple urine test for MPS. I remember bringing the urine to the lab and mostly shrugging it off. It was a busy next few weeks with appointments and then a hospitalization for some scopes and a pH probe. My son had also gotten into a program called the Declan Donoghue Program, this is a program for all specialists to come together to try to find a diagnosis for medically complex children. The next morning after discharge, November 8th 2017, I received a phone call from my son’s Neurology office asking us to come in that afternoon for results. I knew with prior medical experience and almost common sense that this was not good. My husband was hours away and I called my mom who met me at the hospital. 12:00 pm, that was his appointment time and that was the time our lives changed forever. That was the time that our biggest fears came true. MPS, Mucopolysaccharidoses, Probably Hurler (MPS l) or Hunter Syndrome (MPS ll). I busted into tears. Not my baby, I could NOT lose my child. NO cure. Treatment, yes. But even then, this treatment will not reach his brain which in return will not help with neurological decline. I pretty much went through all the stages of grief within 24-48 hours, but the crazy thing was, I had not lost my son yet. He was right in front of me. This diagnosis did not CHANGE who my son was, it was just another obstacle for us to face. The next month or so was a blur. We received confirmation of Hunter Syndrome. Hunter Syndrome or MPS ll is a lysosomal storage disease/metabolic disorder. It is caused by missing or making little enzyme. There are less than 500 cases in the US and 2,000 cases in the world. Because these boys (typically only seen in boys) lack enough of this enzyme, they cannot break down certain complex molecules, which then build up in harmful amounts. Some signs and symptoms include but are not limited to: coarse facial features, bone abnormalities, distended abdomen, diarrhea, delayed development, hydrocephalus, umbilical/inguinal hernia, sleep apnea, airway abnormalities, hearing loss, frequent infections, heart problems, and enlarged liver/spleen. This disease affects every organ in the body and heart failure and respiratory problems are typically the leading causes of death. The life span is typically only 10-15 years old. The neurological aspect mocks that of Alzheimer’s. Typically the boys gain normal skills and then slowly lose the ability to walk, talk & eat. For Easton, unfortunately, he lost his ability to eat as a baby, and he never gained words past mama and dada and even those have come and gone. We are so thankful that he is walking though that has been a journey in itself. Easton has had two airway surgeries, tonsils and adenoids removed, ear tubes placed, and many other anesthesia events. He uses a TLSO back brace, AFO foot braces, high flow oxygen to sleep, a CPT vest to clear mucus from his lungs and is on multiple medications. Easton is and always has been the happiest and most charming little boy. His bravery, strength and intelligence is inspiring. Easton does hours long weekly enzyme treatments which require a port a cath for easy access, he has multiple specialists which has concluded in him gaining a Complex Care team. He does weekly therapies for ST, PT and OT as well. There are multiple procedures, testing, surgeries, etc. required with this diagnosis. Easton LOVES to be a typical little boy and his favorite things are Mickey Mouse/Disney World, swimming, climbing on things, playing with his doggies, spending time with family and riding in his awesome Golf cart from Make A Wish.